Computer assist
First-principles calculations
 VASP and CASTEP are available by using the super computer “TSUBAME” in Tokyo Tech.
We use the program for designing our new materials and also for revealing the underlying mechanism of their functions.
We can freely use the workstations in many cases.
VASP and CASTEP are available by using the super computer “TSUBAME” in Tokyo Tech.
We use the program for designing our new materials and also for revealing the underlying mechanism of their functions.
We can freely use the workstations in many cases.
Molecular dynamics simulations
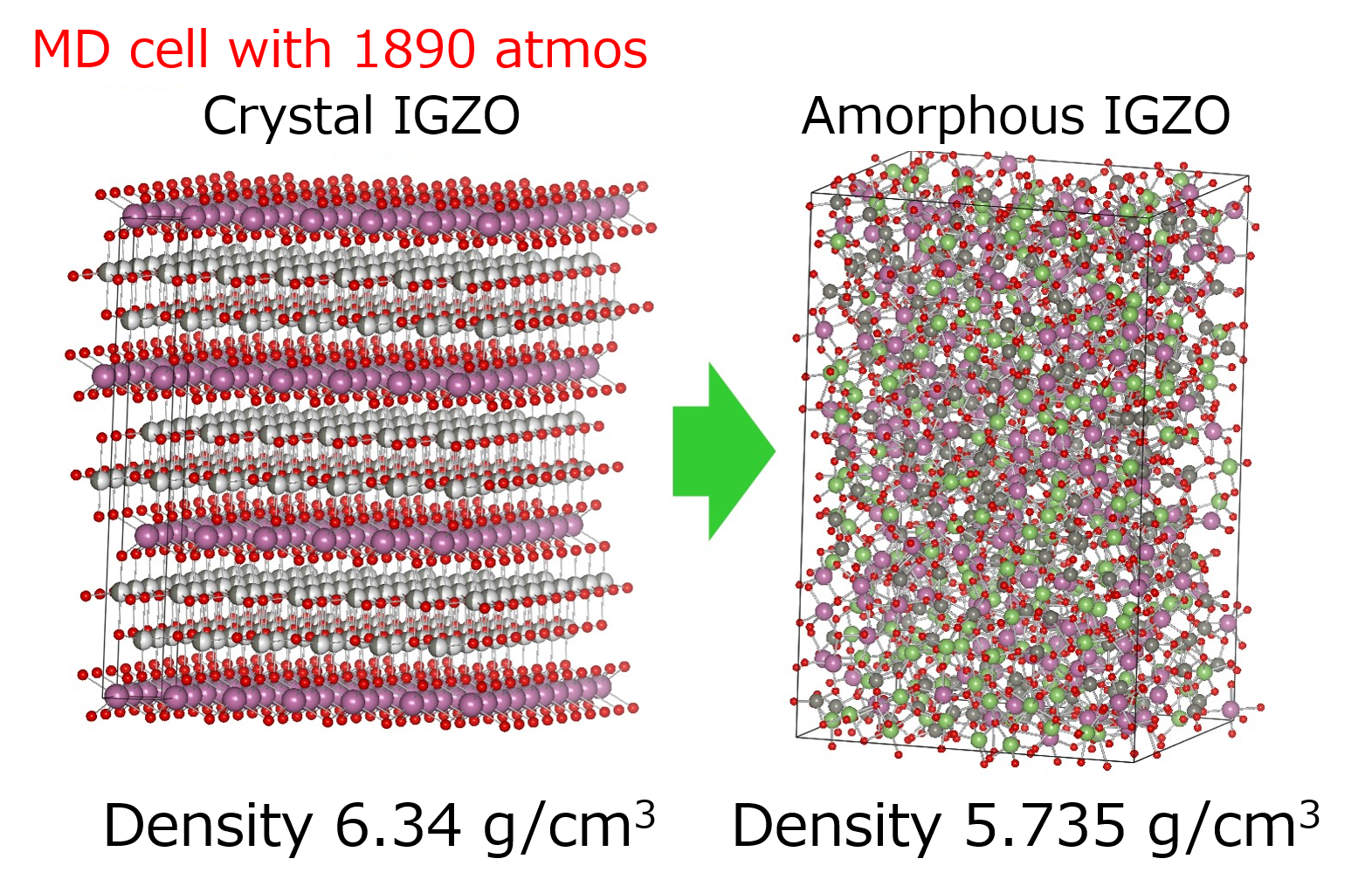 We use Molecular dynamics simulation for revealing the physical movements of atoms and ions in amorphous materials.
The combination with First-principle calculations is a powerful approach to explore new materials.
We use Molecular dynamics simulation for revealing the physical movements of atoms and ions in amorphous materials.
The combination with First-principle calculations is a powerful approach to explore new materials.
Device simulations
ATLAS (Silvaco Inc.) program is available. By defining the defects in semiconductors,
we understand the relationship between device characteristics and the material’s defect levels.
We can also understand the device properties by visualizing the charge density and potential distribution in the devices.
Data simulations
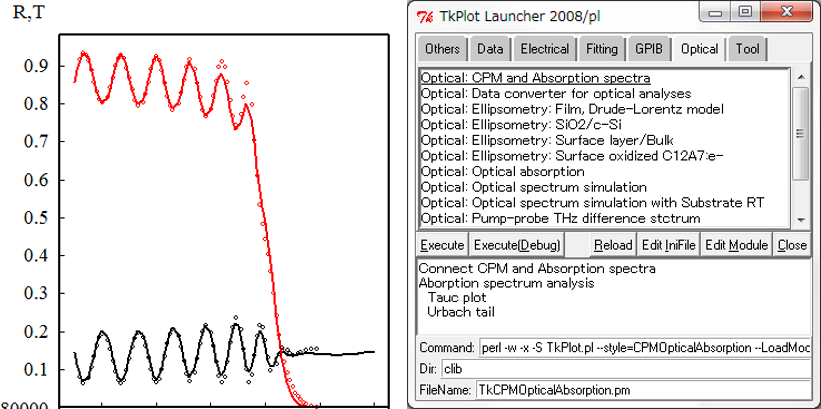 We use our original data simulation system. Numerical analysis is possible for XRD, PES, etc. by least-squares method.
For example, we can easily analyze the dielectric functions and thicknesses from optical spectra.
We use our original data simulation system. Numerical analysis is possible for XRD, PES, etc. by least-squares method.
For example, we can easily analyze the dielectric functions and thicknesses from optical spectra.