Computer assist
コンピュータを使うことにより、実際の材料について、結晶構造、原子配列、電子構造、電子分布、波動関数の形や、半導体に重要な物性がわかるようになっています。新しい材料を創るためには、実験が一番大事ですが、実験をしながら、必要な場合にはコンピュータによる理論計算をして、実験と理論の両面から研究を進める必要があります。当研究室では、分子動力学法、第一原理量子計算、デバイスシミュレータなどを使いながら、研究を進めています。
第一原理計算
 世界的に最も多く用いられているVASPのライセンスおよびCASTEPも使用できます。
東工大はスーパーコンピュータTSUBAMEを有しておりこれを使用することが可能です。
我々の使途は大規模計算ではなく、実験結果の解釈や予測、および知見に基づく材料設計の為に使用します。
そのため多くの場合は研究室内に備えるワークステーションで気軽に計算を実行することが可能です。
世界的に最も多く用いられているVASPのライセンスおよびCASTEPも使用できます。
東工大はスーパーコンピュータTSUBAMEを有しておりこれを使用することが可能です。
我々の使途は大規模計算ではなく、実験結果の解釈や予測、および知見に基づく材料設計の為に使用します。
そのため多くの場合は研究室内に備えるワークステーションで気軽に計算を実行することが可能です。
分子動力学計算
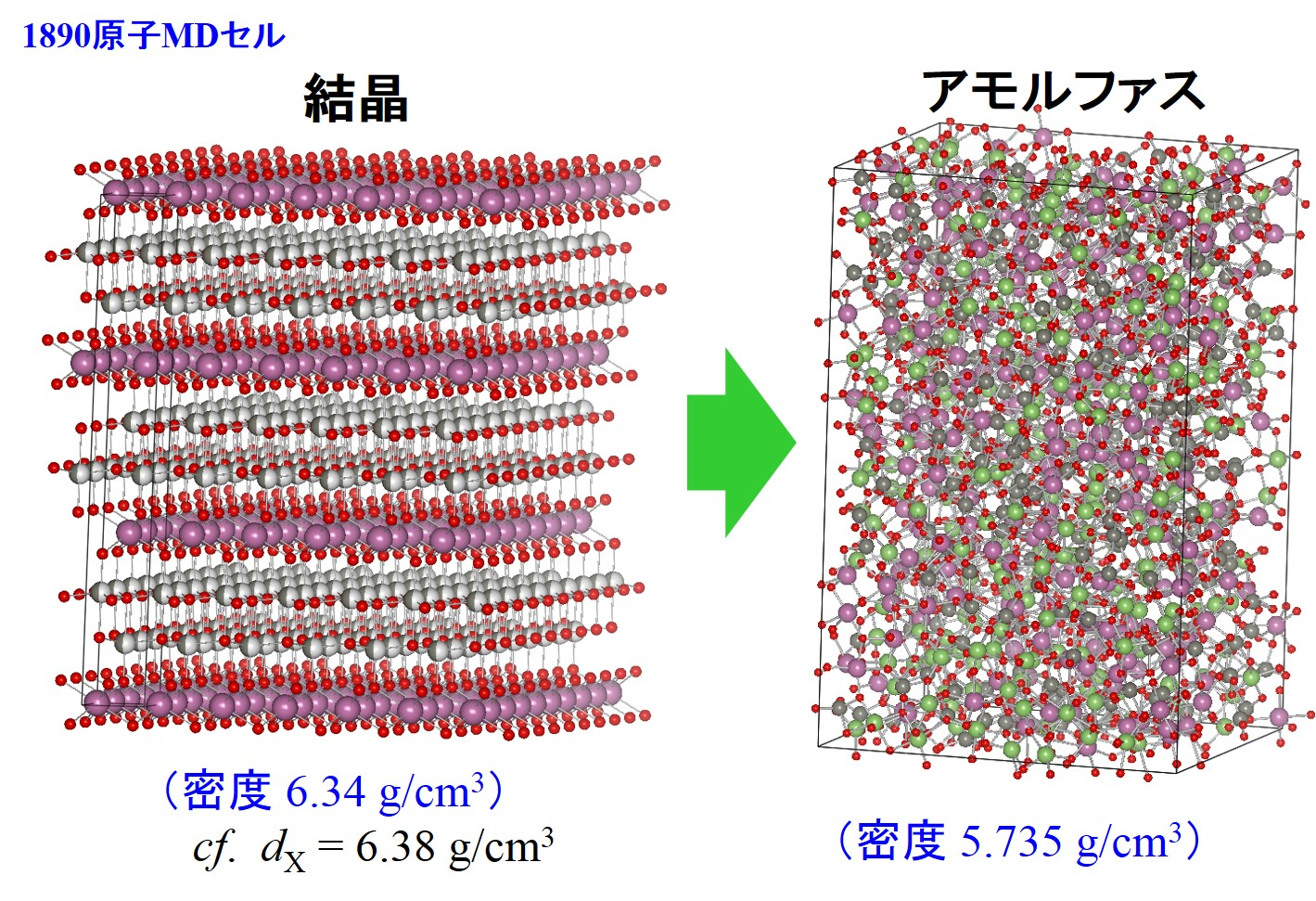 分子動力学計算も特にアモルファス系の計算の際に必要となることが多くあります。
先述のCASTEPや、SCIGRESSのライセンスも保有しいつでも実行可能な環境を整えてあります。
第一原理計算とあわせることで非常に強力な、材料探索のツールとなります。
分子動力学計算も特にアモルファス系の計算の際に必要となることが多くあります。
先述のCASTEPや、SCIGRESSのライセンスも保有しいつでも実行可能な環境を整えてあります。
第一原理計算とあわせることで非常に強力な、材料探索のツールとなります。
デバイスシミュレーション
 Silvaco社のATLASを使用しています。
これを使って半導体の欠陥を定義することで、デバイス特性と材料の欠陥準位を対応させることができます。
またデバイス内でのポテンシャル分布、電荷密度を計算し可視化できるため、容易にデバイス動作を理解することが出来ます。
Silvaco社のATLASを使用しています。
これを使って半導体の欠陥を定義することで、デバイス特性と材料の欠陥準位を対応させることができます。
またデバイス内でのポテンシャル分布、電荷密度を計算し可視化できるため、容易にデバイス動作を理解することが出来ます。
データ解析
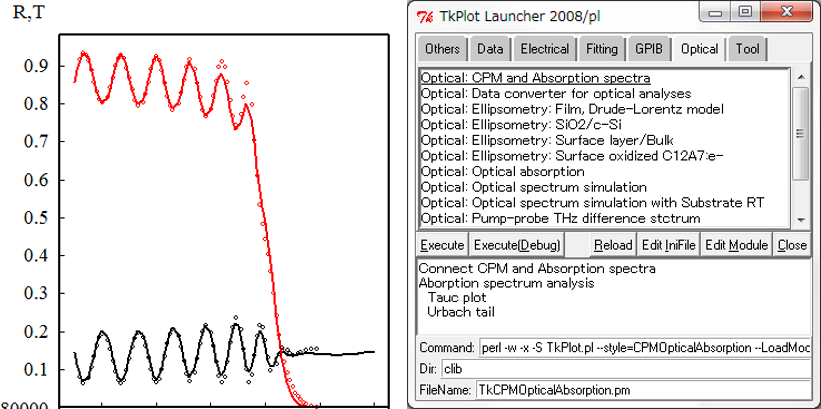 右図は我々オリジナルの解析プログラムです。最小二乗法を駆使したあらゆるデータ解析に使用することができます。
特に光学スペクトルの解析のような沢山のパラメータが存在する場合に有効で、膜厚・誘電関数を簡単に算出することができます。
他にも光電子分光やX線回折や数値処理に使うことが出来ます。
右図は我々オリジナルの解析プログラムです。最小二乗法を駆使したあらゆるデータ解析に使用することができます。
特に光学スペクトルの解析のような沢山のパラメータが存在する場合に有効で、膜厚・誘電関数を簡単に算出することができます。
他にも光電子分光やX線回折や数値処理に使うことが出来ます。